Permutation score analysis - Spermatogenesis#
Library imports#
import os
import sys
import numpy as np
import pandas as pd
import torch
from scipy import stats
from sklearn.preprocessing import MinMaxScaler
import matplotlib.pyplot as plt
import mplscience
import seaborn as sns
import scanpy as sc
import scvelo as scv
import scvi
from scvelo.plotting.simulation import compute_dynamics
from velovi import VELOVI
sys.path.append("../..")
from paths import DATA_DIR, FIG_DIR
Global seed set to 0
General settings#
scvi.settings.dl_pin_memory_gpu_training = False
sns.reset_defaults()
sns.reset_orig()
scv.settings.set_figure_params('scvelo', dpi_save=400, dpi=80, transparent=True, fontsize=20, color_map='viridis')
SAVE_FIGURES = True
if SAVE_FIGURES:
os.makedirs(FIG_DIR / 'permutation' / 'spermatogenesis', exist_ok=True)
Function definitions#
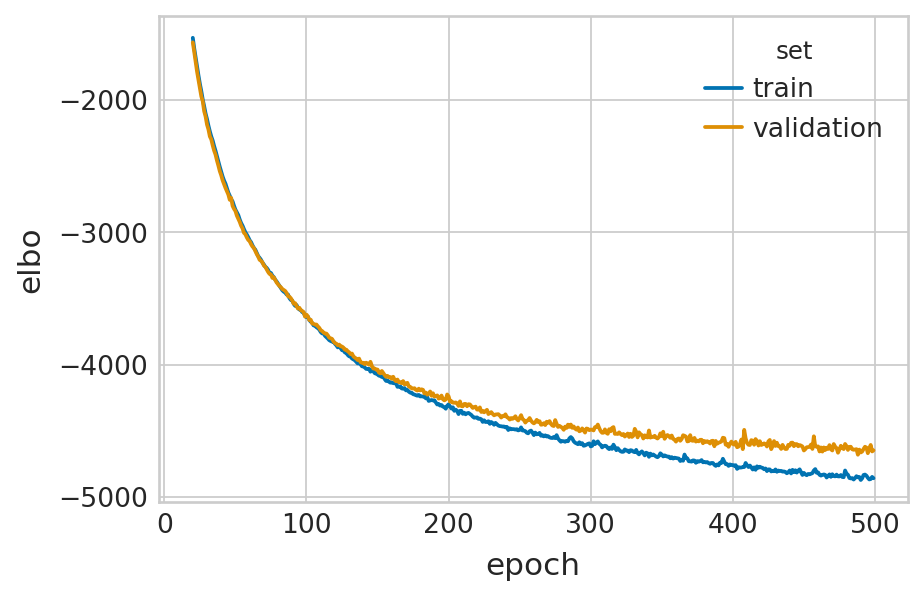
def fit_velovi(bdata):
VELOVI.setup_anndata(bdata, spliced_layer="Ms", unspliced_layer="Mu")
vae = VELOVI(bdata)
vae.train()
df = vae.history["elbo_train"].iloc[20:].reset_index().rename(columns={'elbo_train': 'elbo'})
df['set'] = 'train'
_df = vae.history["elbo_validation"].iloc[20:].reset_index().rename(columns={'elbo_validation': 'elbo'})
_df['set'] = 'validation'
df = pd.concat([df, _df], axis=0).reset_index(drop=True)
with mplscience.style_context():
sns.set_style(style="whitegrid")
fig, ax = plt.subplots(figsize=(6, 4))
sns.lineplot(data=df, x='epoch', y='elbo', hue='set', palette=['#0173B2', '#DE8F05'], ax=ax)
latent_time = vae.get_latent_time(n_samples=25)
velocities = vae.get_velocity(n_samples=25, velo_statistic="mean")
t = latent_time
scaling = 20 / t.max(0)
bdata.layers["velocities_velovi"] = velocities / scaling
bdata.layers["latent_time_velovi"] = latent_time
bdata.var["fit_alpha"] = vae.get_rates()["alpha"] / scaling
bdata.var["fit_beta"] = vae.get_rates()["beta"] / scaling
bdata.var["fit_gamma"] = vae.get_rates()["gamma"] / scaling
bdata.var["fit_t_"] = (
torch.nn.functional.softplus(vae.module.switch_time_unconstr)
.detach()
.cpu()
.numpy()
) * scaling
bdata.layers["fit_t"] = latent_time.values * scaling[np.newaxis, :]
bdata.var['fit_scaling'] = 1.0
return vae
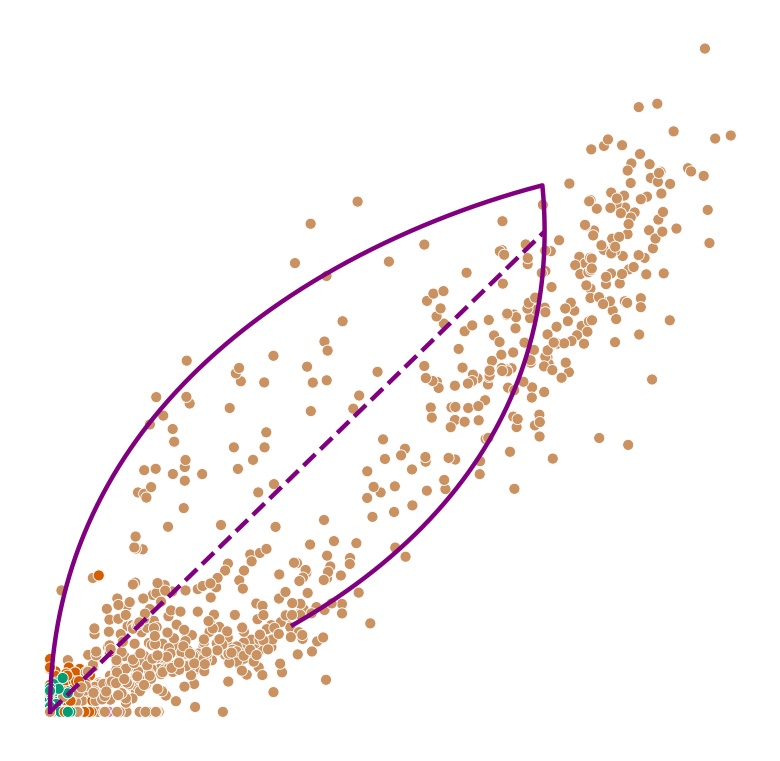
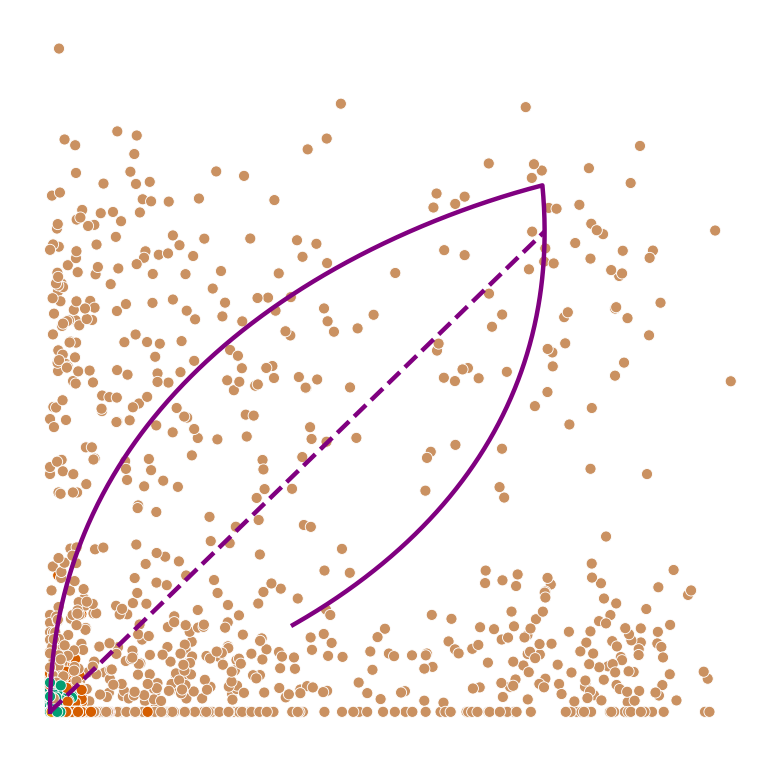
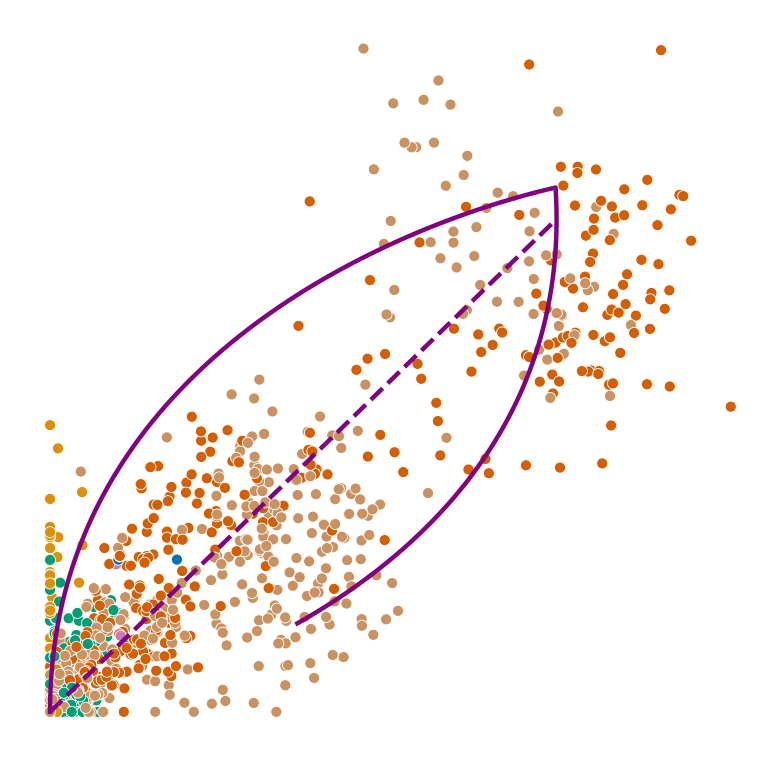
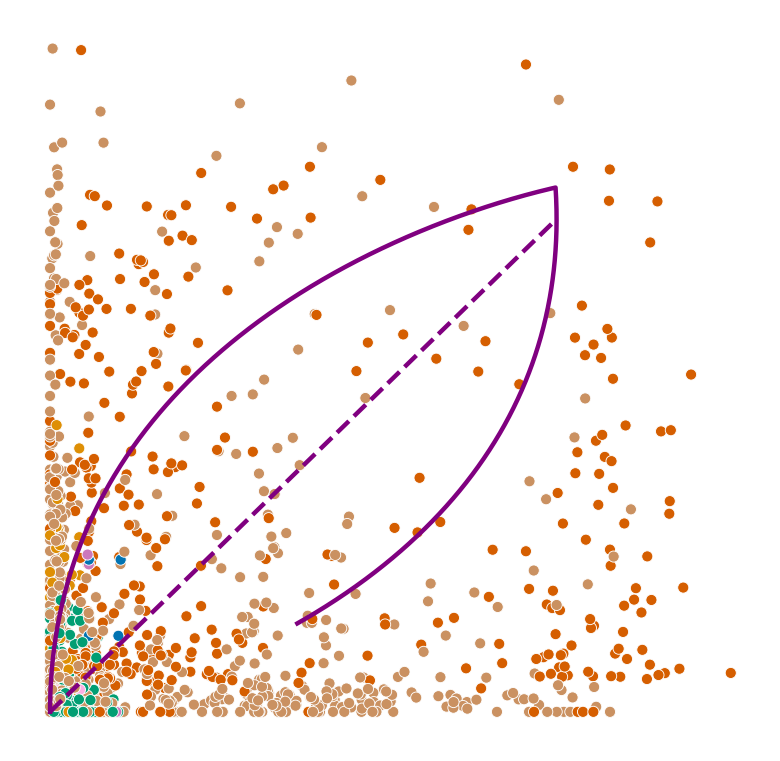
def plot_phase_portrait(adata, gene, color, permuted=False, figsize=(6, 6)):
fig, ax = plt.subplots(figsize=figsize)
df = pd.DataFrame(
{
'unspliced': adata[:, gene].layers['Mu'].squeeze().copy(),
'spliced': adata[:, gene].layers['Ms'].squeeze().copy(),
'color': color
}
)
with mplscience.style_context():
sns.scatterplot(data=df, x='spliced', y='unspliced', c=color, s=25, ax=ax);
_, unspliced, spliced = compute_dynamics(adata, basis=gene, extrapolate=True, sort=True)
df = pd.DataFrame(
{
'unspliced': unspliced.squeeze(),
'spliced': spliced.squeeze(),
}
)
ax.plot(spliced, unspliced, color="purple", linewidth=2)
spliced_steady_state = np.linspace(np.min(spliced), np.max(spliced))
unspliced_steady_state = adata.var.loc[gene, 'fit_gamma'] / adata.var.loc[gene, 'fit_beta'] * (spliced_steady_state - np.min(spliced_steady_state)) + np.min(unspliced)
ax.plot(spliced_steady_state, unspliced_steady_state, color='purple', linestyle="--", linewidth=2);
ax.axis('off')
if SAVE_FIGURES:
if permuted:
fname = f'phase_portrait_{gene}_permuted'
else:
fname = f'phase_portrait_{gene}'
fig.savefig(
FIG_DIR / 'permutation' / 'spermatogenesis' / f'{fname}.svg',
format="svg",
transparent=True,
bbox_inches='tight'
)
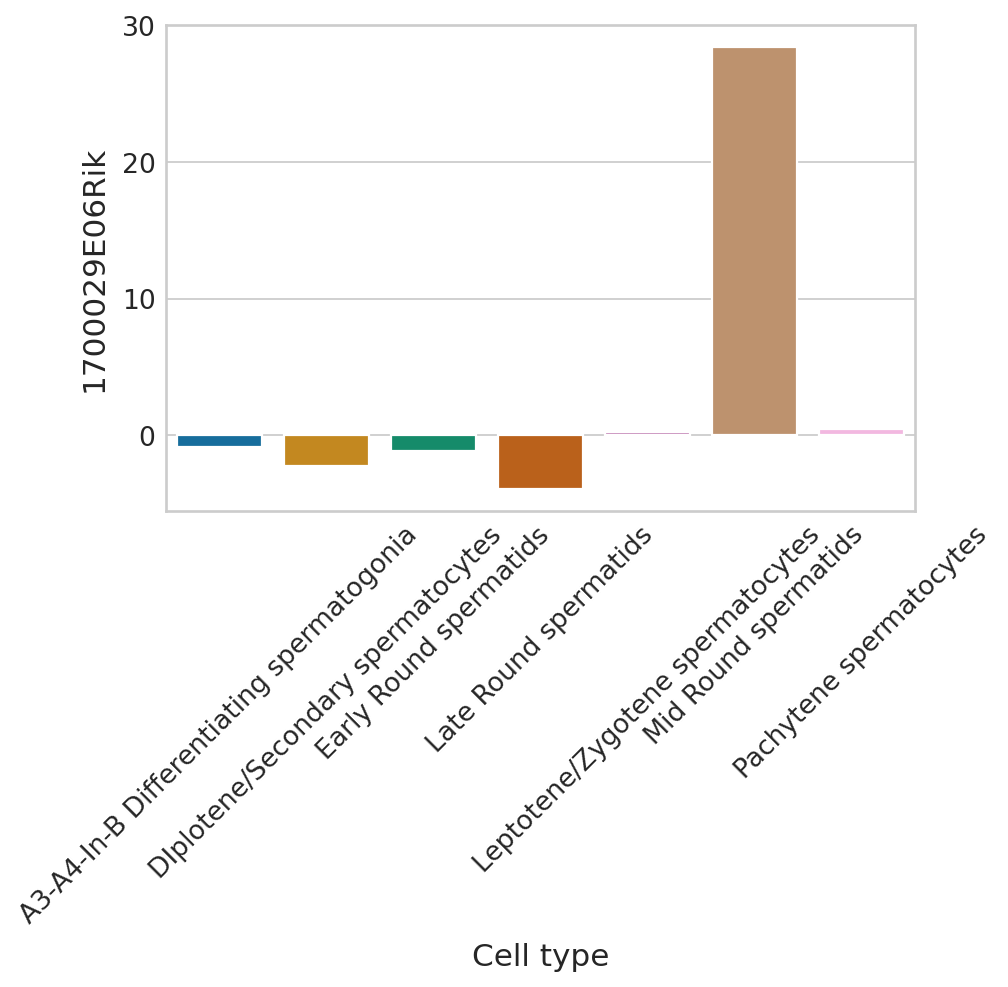
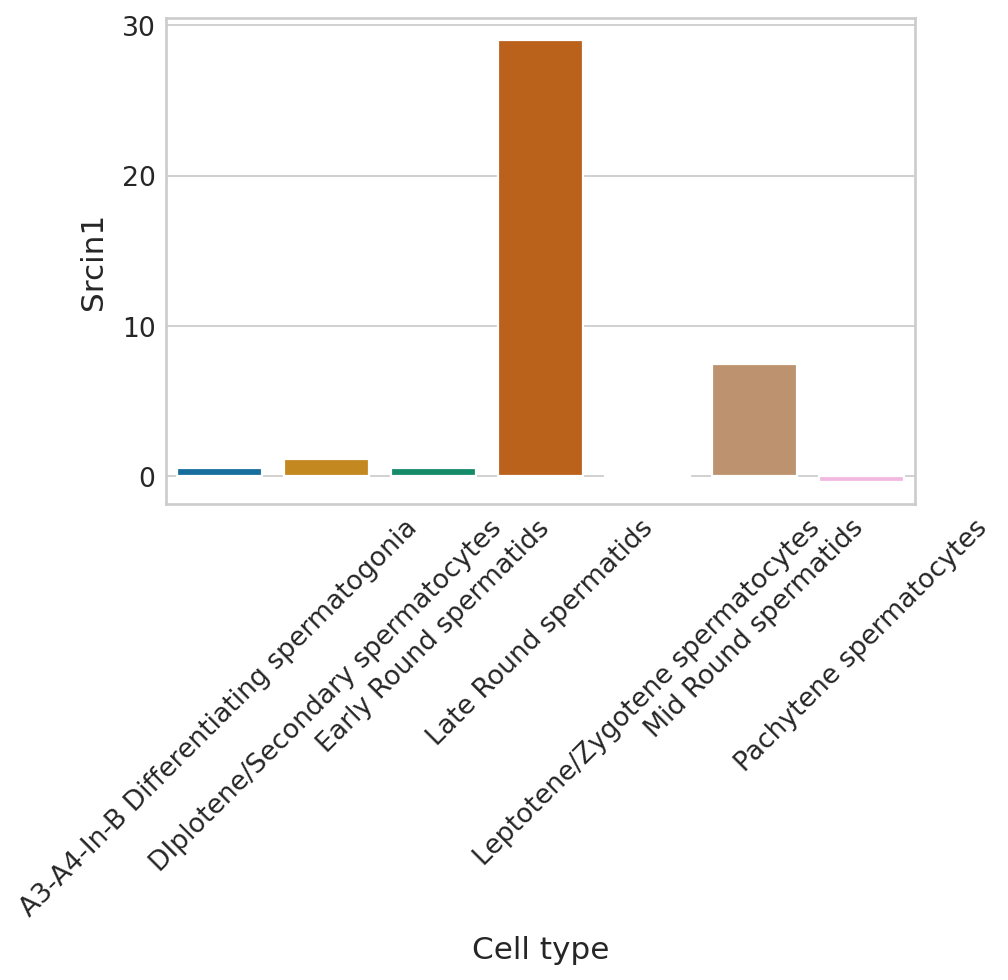
def plot_perm_scores(adata, perm_scores, gene, color_label, figsize=(6, 4)):
df = pd.DataFrame(perm_scores.loc[gene])
df["Cell type"] = df.index
order = adata.obs[color_label].cat.categories.tolist()
with mplscience.style_context():
sns.set_style(style="whitegrid")
fig, ax = plt.subplots(figsize=figsize)
sns.barplot(
data=df,
y=gene,
x="Cell type",
palette=adata.uns[f"{color_label}_colors"],
order=order,
ax=ax,
)
ax.tick_params(axis='x', rotation=45)
if SAVE_FIGURES:
fig.savefig(
FIG_DIR / 'permutation' / 'spermatogenesis' / f'permutation_score_{gene}.svg',
format="svg",
transparent=True,
bbox_inches='tight'
)
Data loading#
adata = sc.read(DATA_DIR / "spermatogenesis" / "spermatogenesis.h5ad")
adata.uns['clusters_colors'] = sns.color_palette('colorblind').as_hex()[:len(adata.obs['clusters'].unique())]
adata
AnnData object with n_obs × n_vars = 1829 × 54144
obs: 'cell_index', 'clusters_coarse', 'clusters'
uns: 'clusters_colors'
layers: 'spliced', 'unspliced'
Data preprocessing#
scv.pp.filter_and_normalize(adata, min_shared_counts=20, n_top_genes=2000)
scv.pp.moments(adata, n_pcs=30, n_neighbors=30)
scaler = MinMaxScaler()
adata.layers["Mu"] = scaler.fit_transform(adata.layers["Mu"])
scaler = MinMaxScaler()
adata.layers["Ms"] = scaler.fit_transform(adata.layers["Ms"])
Filtered out 46556 genes that are detected 20 counts (shared).
Normalized count data: X, spliced, unspliced.
Extracted 2000 highly variable genes.
Logarithmized X.
computing neighbors
finished (0:00:05) --> added
'distances' and 'connectivities', weighted adjacency matrices (adata.obsp)
computing moments based on connectivities
finished (0:00:00) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
Model fitting#
velovi_vae = fit_velovi(adata)
/home/icb/philipp.weiler/miniconda3/envs/velovi-py39/lib/python3.9/site-packages/torch/distributed/_sharded_tensor/__init__.py:8: DeprecationWarning: torch.distributed._sharded_tensor will be deprecated, use torch.distributed._shard.sharded_tensor instead
warnings.warn(
GPU available: True, used: True
TPU available: False, using: 0 TPU cores
IPU available: False, using: 0 IPUs
LOCAL_RANK: 0 - CUDA_VISIBLE_DEVICES: [0]
Set SLURM handle signals.
Epoch 500/500: 100%|██████████| 500/500 [01:15<00:00, 6.64it/s, loss=-4.86e+03, v_num=1]
Permutation score evaluation#
perm_scores, permuted_adata = velovi_vae.get_permutation_scores(labels_key='clusters')
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
INFO Input AnnData not setup with scvi-tools. attempting to transfer AnnData setup
full_perm_df = pd.DataFrame(columns=["Score", "Dataset"])
max_ratio = np.nanmax(perm_scores.values, axis=1)
scores = max_ratio.tolist()
dataset = ['Spermatogenesis'] * len(max_ratio)
full_perm_df["Score"] = scores
full_perm_df["Dataset"] = dataset
color = adata.obs['clusters'].astype(str).replace(
dict(zip(adata.obs['clusters'].cat.categories, adata.uns['clusters_colors']))
).tolist()
Lowest ranked genes#
for gene in adata.var_names[np.argsort(scores)[:2]]:
plot_phase_portrait(adata, gene, color)
plot_phase_portrait(permuted_adata, gene, color, permuted=True)
plot_perm_scores(adata, perm_scores, gene, 'clusters')
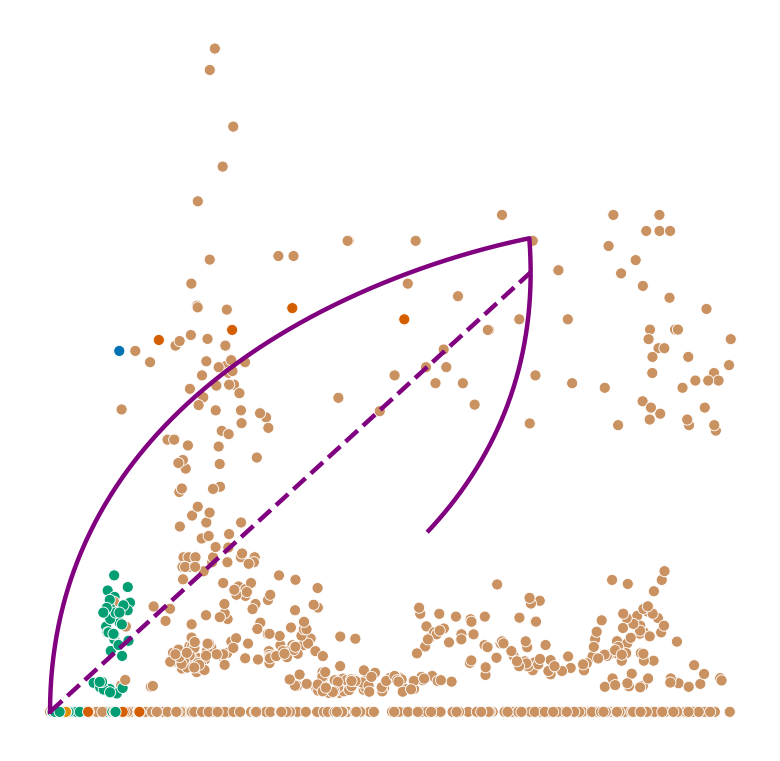
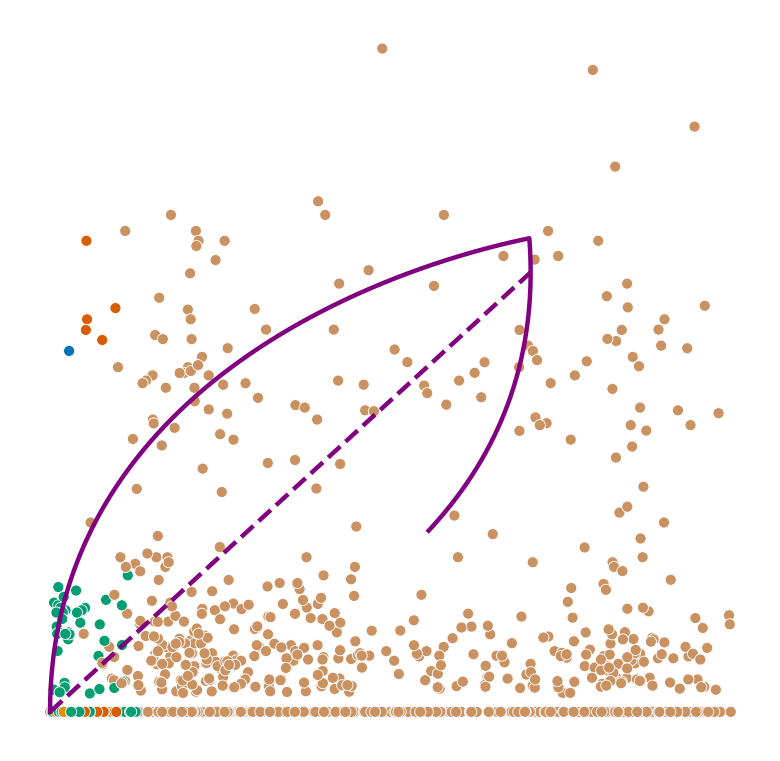
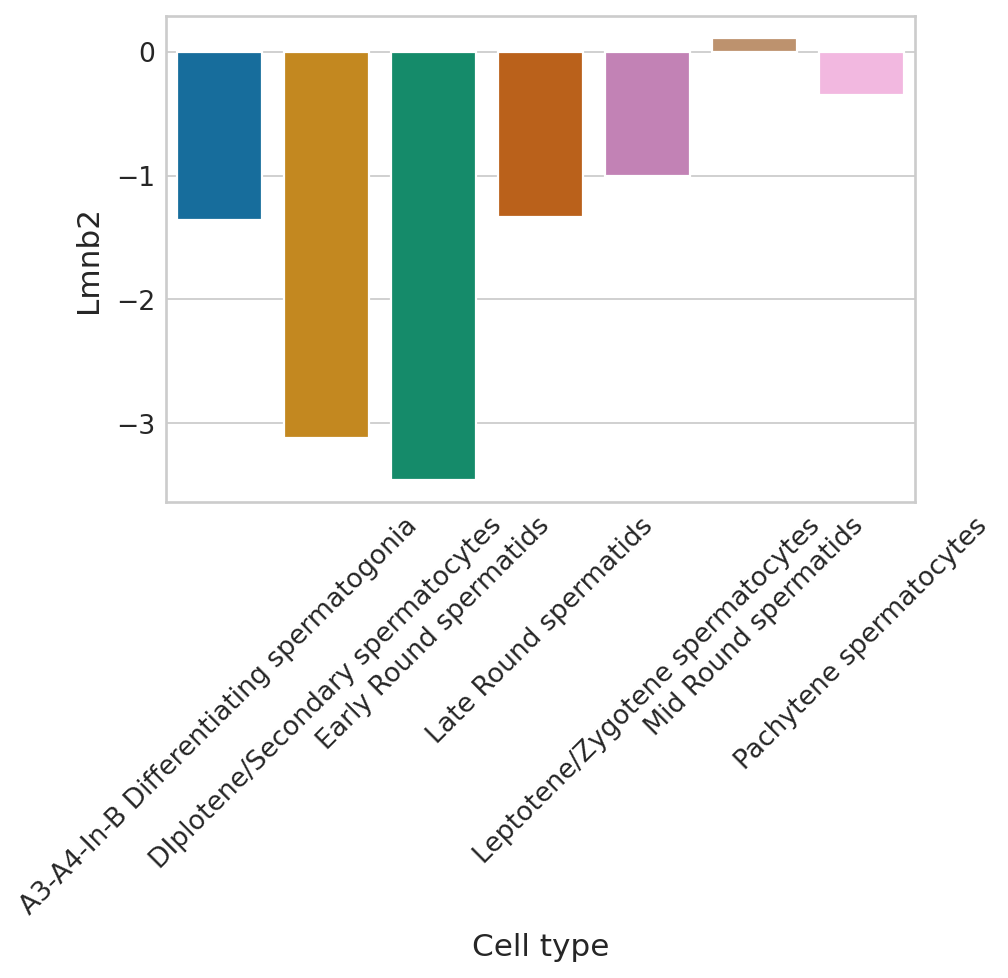
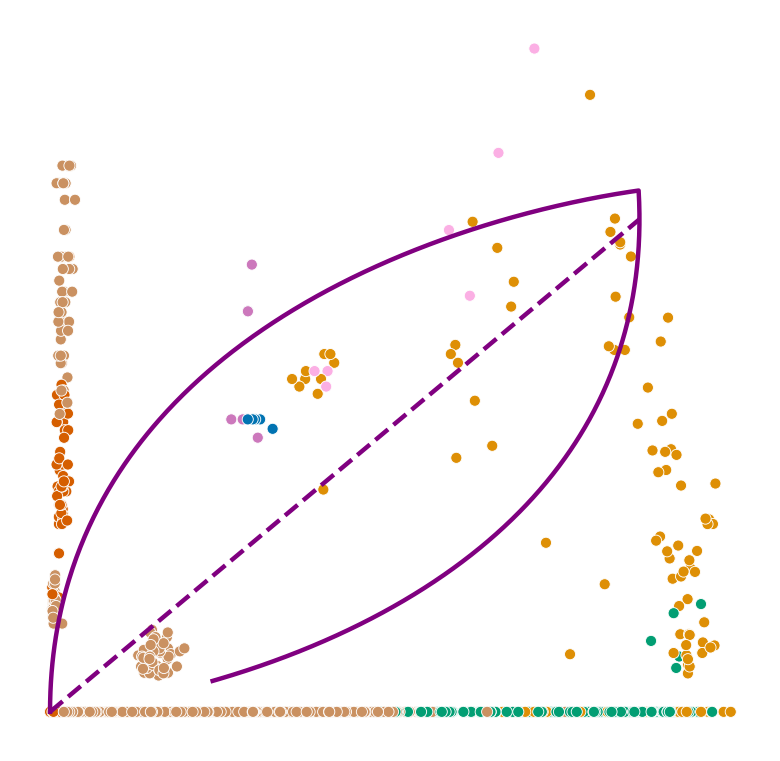
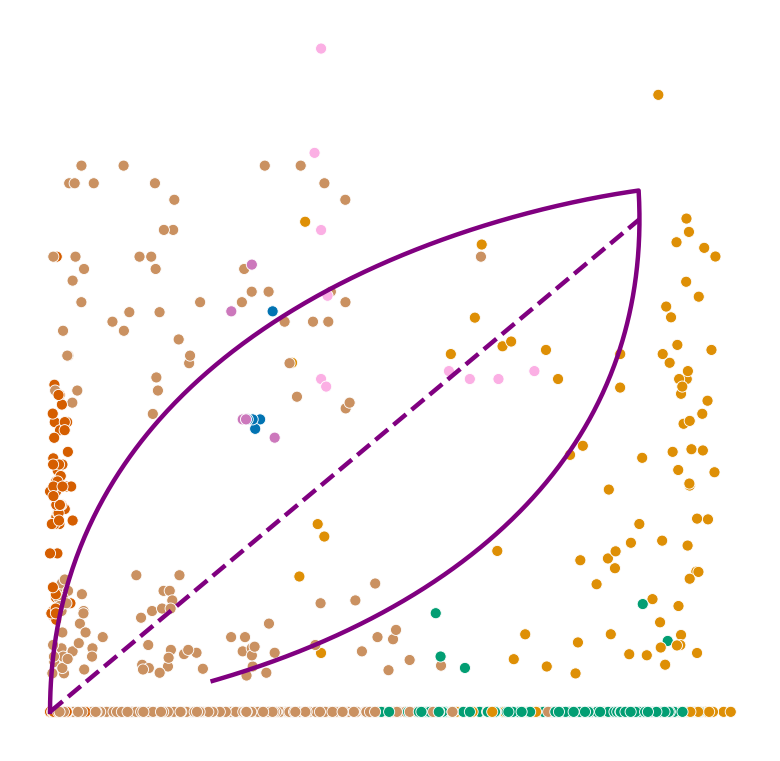
Highest ranked genes#
for gene in adata.var_names[np.argsort(scores)[-2:]]:
plot_phase_portrait(adata, gene, color)
plot_phase_portrait(permuted_adata, gene, color, permuted=True)
plot_perm_scores(adata, perm_scores, gene, 'clusters')