scone
Framework for the evaluation of scRNA-seq normalization
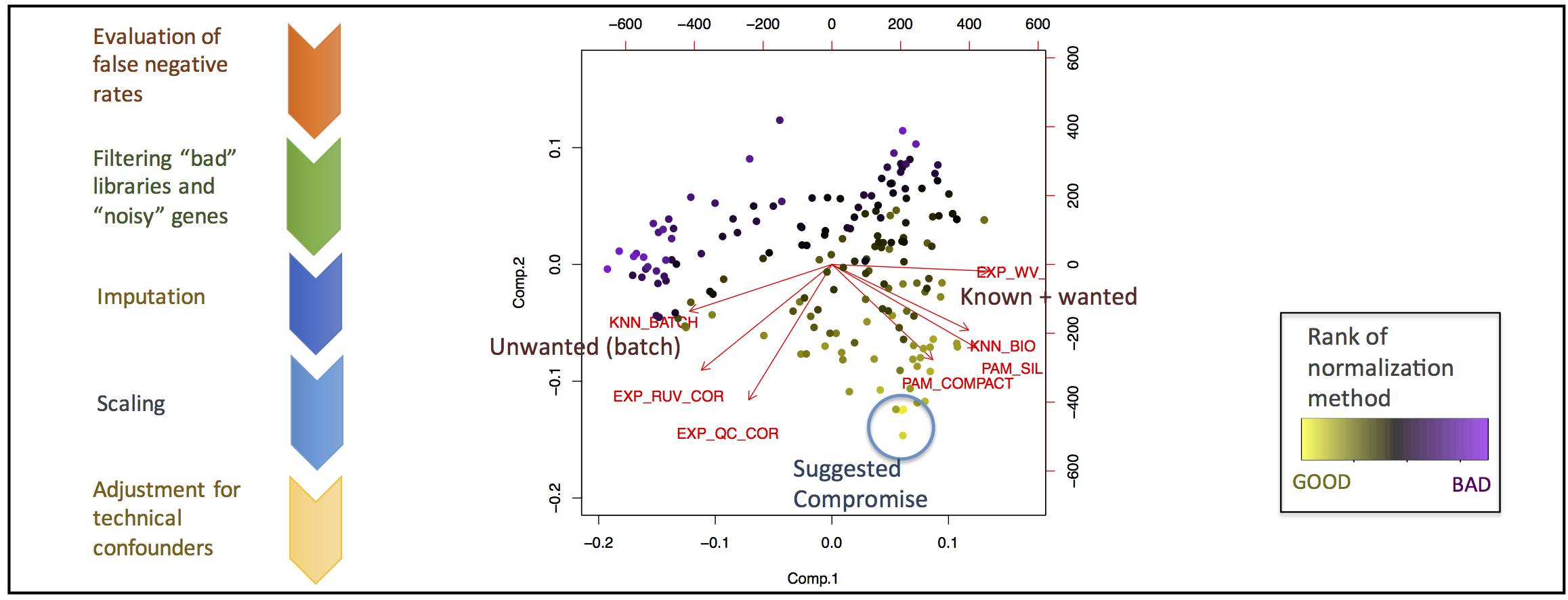
SCONE (Single-Cell Overview of Normalized Expression) is an R Biodconductor package that supports a rational, data-driven framework for assessing the efficacy of various normalization workflows, encouraging users to explore trade-offs inherent to their data set prior to finalizing a data normalization strategy. We provide an interface for running multiple normalization workflows in parallel. We also offer tools for ranking workflows and visualizing trade-offs. We import some common normalization modules used in traditional bulk sequencing, and provide support for integrating user-specified normalization modules.
Input Data
- Expression Matrix (e.g. Read Counts)
- Library Alignment Metrics
- Biological Exposures
- Batch Conditions
- Control Gene Sets
General Normalization Workflow
- Data Imputation Module: replacing zero-abundance values with expected values under a drop-out model.
- Scaling or Quantile Normalization Module: either i) normalization that scales each sample’s transcriptome abundances by a single factor or ii) more complex offsets that match quantiles across samples.
- Regression Module. Approaches for removing unwanted correlated variation from the data (e.g. RUVg, Risso et al. 2014).
Output
- Hundreds of Normalized Expression Matrices
- Up to 8 Performance Metrics per Matrix
- Ranking by Performance Scores
Availability
Download the R Bioconductor package, or checkout the project on GitHub.
Vignette is available here.